
Introduction
Familial atypical multiple mole melanoma (FAMMM) syndrome is an autosomal dominant inherited disorder in which patients have a large number of melanocytic naevi and one or more first or second-degree relatives with malignant melanoma. The number of naevi is usually greater than 50.3 Although 7–15 per cent of patients diagnosed with melanoma report a positive family history, only 1–2 per cent of these patients have FAMMM syndrome.4,5 Patients with FAMMM syndrome are distinct from the majority of melanoma patients, whose melanomas arise due to a combination of sporadic, polygenic and modifiable factors.
The phenotype of the FAMMM syndrome patient differs from the average melanoma patient due to a greater number of malignant melanomas and an earlier age of first melanoma. Their family history shows a particular pattern of malignant melanoma that is multigenerational and of unilateral, autosomal dominant inheritance.6 The most common genetic alteration implicated in FAMMM syndrome is a mutation of cyclin-dependent kinase inhibitor 2A (CDKN2A), which is a tumour suppressor gene involved in cell cycle inhibition.2 CDKN2A alteration is present in approximately 40 per cent of families diagnosed with FAMMM syndrome.1,3,6 FAMMM syndrome is associated with but distinct to several syndromes, such as other forms of hereditary melanoma and B-K mole syndrome.
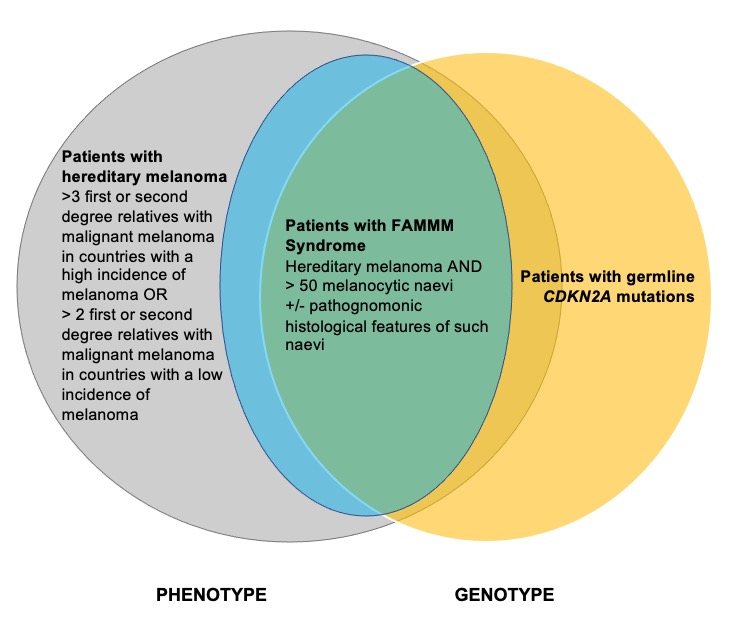
The diagnosis and management of patients with FAMMM syndrome is relevant to the plastic surgeon who manages melanoma. However, clear guidelines on its diagnostic criteria and management are lacking in the literature. This review has three objectives: first, to clarify the current diagnostic criteria for FAMMM syndrome; second, to describe a system of classifying FAMMM syndrome patients as a subset of all patients with hereditary melanoma (illustrated in Figure 2); and third, to present a management algorithm that differentiates patients with FAMMM syndrome, hereditary melanoma and CDKN2A mutations.
__he.jpg)
The historical context of FAMMM syndrome
The diagnostic criteria for FAMMM syndrome have evolved over time, from the description of B-K mole syndrome to the discovery of the CDKN2A gene on chromosome 9p21. The first description of inherited melanoma occurring in a patient with multiple pigmented naevi was in 1820 by Norris.7 His description was of a 59-year-old man with multiple pigmented naevi, a few of which had developed into recurrent, invasive metastatic melanoma. The patient’s children, father and brothers also had an unusually large number of pigmented naevi.7 The next description was in 1968, following the emergence of molecular genetics. Lynch and Krush published a case series of four families with high rates of malignant melanoma. Through mapping the family pedigrees of their probands, they deduced a genetic disorder of inherited melanoma. They described a syndrome of inherited melanoma presenting with multiple primary melanomas, early age of first melanoma and autosomal dominant inheritance with variable penetrance.8
Lynch and Krush first defined FAMMM syndrome. They described these patients as having ‘multiple large moles, irregular in shape, coloured reddish-brown to pink, with evidence of pigmentary leakage’. Such moles had a high risk of developing into melanomas years later and were typically located on non-sun-exposed areas such as the chest and back. After pedigree mapping a number of affected families, Lynch and Krush discerned a pattern of direct transmission from parent to progeny and equal sex distribution, suggestive of autosomal dominant inheritance.9 Cases in which the FAMMM syndrome appeared to skip a generation were explained by incomplete penetrance.10,11
In 1978, Clark and colleagues described ‘B-K mole syndrome’, a genetic syndrome in which patients presented with a histologically unique type of pigmented naevus that could develop into melanoma. The diagnosis was based on certain histologic features of the naevus, such as atypical melanocytic hyperplasia, lymphocytic infiltration, delicate fibroplasia and the formation of new blood vessels. They defined two subtypes of the syndrome: one where the individual presents with fewer than 10 pigmented naevi, and the other where the individual presents with greater than 100 pigmented naevi.12
Lynch and colleagues’s historical diagnostic criteria for FAMMM syndrome did not include specific histological features, while Clark’s diagnostic criteria for B-K mole syndrome did.10,12 Other names for FAMMM syndrome include ‘large atypical mole syndrome’, ‘expanded and activated melanocyte syndrome’ and ‘dysplastic nevus syndrome’.13–15 Lynch and colleagues also emphasised two crucial aspects of the disease. First, that families with FAMMM syndrome have a higher incidence of pancreatic cancer. And second, that patients with FAMMM syndrome may present atypically—without multiple melanocytic naevi, but still experience de novo occurrences of melanoma.10
Today, the Genetic and Rare Diseases Information Center (GARD) and Orphanet define FAMMM syndrome as an autosomal dominant genetic condition in which the following three criteria must be met. Within this article, the term ‘FAMMM syndrome’ refers to patients who meet these particular criteria:
- One or more first or second-degree relatives with malignant melanoma
- Greater than 50 melanocytic naevi, some of which are atypical and often with a large variability in size
- Melanocytic nevi characterised by the following histologic features on microscopy:
a. architectural disorder with asymmetry
b. subepidermal fibroplasia
c. lentiginous melanocytic hyperplasia with spindle or epithelioid melanocytes gathering in nests of variable size and fusing with adjacent rete ridges to form bridges
d. variable dermal lymphocyte infiltration
e. intra-epithelial melanocytes extending beyond the main dermal component (‘shouldering’ phenomenon).3,16,17
The discovery of CDKN2A
With the rise of genetic linkage analysis and positional cloning techniques in the early 1990s, evidence grew to suggest a specific melanoma susceptibility locus on chromosome 9p21. It was identified as the most frequently deleted region of the human genome in melanoma.18 Moreover, in families with inherited melanomas, a range of DNA polymorphisms near the 9p region were shown to segregate with disease, suggesting that this region might be the locus for a tumour suppressor gene.18,19 Patients from high-risk melanoma kindreds who had inherited the disease-linked 9p21 allele were found to have a greater number of naevi, a greater naevus density, a lighter skin pigmentation and a higher than expected incidence of melanoma, compared to non-carriers.19
Shortly afterwards, the gene implicated on chromosome 9p21 was identified as the CDKN2A gene, which encodes the protein p16INK4A (p16) and acts as a tumour suppressor gene in line with Knudson’s two-hit hypothesis.20,21 As shown in Figure 1, CDKN2A is comprised of four exons—1α, 1β, 2 and 3—and encodes two distinct proteins translated from alternatively spliced transcripts. Exons 1α, 2 and 3 encode the protein p16, which binds to cyclin-dependent kinase 4 (CDK4), preventing interaction with cyclin D and inhibiting phosphorylation of the retinoblastoma protein (Rb) and thus passage from the G1 to S phases of the cell cycle. Exons 1β, 2 and 3 encode the protein p14ARF (p14), which stabilises p53 via inhibition of MDM2-induced p53 degradation. Mutations in CDKN2A may thus lead to impaired regulation of the cell cycle and the unfettered proliferation of aberrant cells.20–24 It has since been identified that CDKN2A mutations that affect p16 are usually loss-of-function missense mutations; however, nonsense, promotor, frameshift, in-frame insertion, splicing and gene deletion have also been reported.25,26 Mutations that inactivate p14 are most commonly splice site variants,25,27 inactivating frameshift mutations28 or exon 1β-specific deletions.29

Hereditary melanoma
Patients with hereditary melanoma are those that have a personal and family history of melanoma which warrants genetic testing; those that warrant genetic testing do so because the pre-test probability of identifying a pathogenic mutation is greater than or equal to 10 per cent.32 While 7–15 per cent of melanoma patients report a positive family history, only 5–10 per cent have true hereditary melanoma and only 2 per cent of melanoma patients have an identifiable pathogenic germline CDKN2A mutation.4,33,34 Genetic testing for CDKN2A mutations first became available in the mid-2000s. At first, its use in clinical practice was controversial due to the variability in the penetrance of disease in CDKN2A mutation carriers, estimated to be between 13 and 91 per cent.35–38
In 2009, Leachman and colleagues analysed available published studies of CDKN2A mutation analysis in cohorts with cutaneous melanoma. Based on their results, they formulated a decision aid for clinicians for identifying patients who are appropriate candidates for genetic consultation and possible testing. The decision aid is presented in Table 1; two separate criteria are presented depending on whether the patient lives in an area of low or high melanoma incidence. Patients who meet the criteria from Table 1 should be considered to have hereditary melanoma.34,39 The risk of melanoma in a patient with a pathogenic CDKN2A mutation may be compounded by a patient’s pigmentation characteristics (having red hair), naevus phenotypes and whether or not they possess one or more pathogenic variants of the melanocortin-1 receptor gene (MC1R).38,40,41
For the most part, patients who meet the criteria from Table 1 have a pre-test probability of testing positive for CDKN2A mutations of approximately 10 per cent.34,39 The main exception to this rule is the Australian population, which has the highest incidence of melanoma in the world, a rate of 54 cases per 100,000 persons in 2020.42 Here, mutation prevalence rates do not increase above 10 per cent until there are at least five cases of melanoma in the family.34 In order to address this, two statistical models (MELPREDICT and MelaPRO) have been created to assist with estimating pre-test probabilities in the Australian population.43,44 More recently, the Familial Risk Assessment of Melanoma (FRAMe) model was created based on data from Australian families. This is the first prediction tool validated using Australian data. It accounts for family variables such as number of individuals diagnosed with melanoma under age 40 and number of individuals diagnosed with more than one primary melanoma.45 FRAMe is available at https://www.melanomarisk.org.au/.
Patients with germline CDKN2A mutations are more likely to have multiple family members with melanoma, more likely to have an earlier median age of diagnosis (less than age 40 in an Australian population)46 and more likely to have family members with multiple primary melanomas or pancreatic cancer. It was later found that only CDKN2A mutations affecting p16 are associated with pancreatic cancer and mutations affecting p14 are associated with a greater risk of neural system tumours, uterine cancer, glioblastoma multiforme and non-Hodgkin’s lymphoma.47,48 Interestingly, no relationship has been found between CDKN2A mutations and pancreatic cancer in the Australian population.1,45 This may reflect a divergent spectrum of mutations in Australian families, compared to families in Europe and North America.1
CDKN2A and other genetic mutations related to melanoma
Since the discovery of CDKN2A, multiple other genetic mutations responsible for melanoma susceptibility have been identified. These include genetic alterations of CDK4 (cyclin-dependent kinase 4),49–51 ACD,52 BAP1 (breast cancer-associated protein-1),53–55 MITF (micropthalmia-associated transcription factor),56–58 POT1 (protection of telomeres 1),59,60 TERF2IP (telomeric repeat-binding factor 2-interacting protein 1)52 and the TERT promoter (telomerase reverse transcriptase).61,62 However, compared to CDKN2A-p16 mutations, which account for approximately 40 per cent of kindreds with inherited melanoma and 2 per cent of all melanoma cases, these subsequently discovered melanoma-predisposing mutations are all relatively rare, accounting for less than 1 per cent of hereditary melanoma.25,63
CDKN2A mutations are implicated in only 20–40 per cent of families with hereditary melanoma; indeed, for 55–80 per cent of patients with hereditary melanoma, no singular causative genetic mutation has been found.1,32,64 In some proposed testing algorithms, patients who meet the criteria in Table 1 and test negative for CDKN2A mutations may be offered further genetic testing. Second-line testing for CDK4 mutations has been recommended, even though such mutations are rare and have been identified in less than 20 families to date.63 More recently, the advent of reasonably priced multi-gene panel tests has made it possible to offer hereditary melanoma patients a one-off genetic panel test for CDKN2A, CDK4, BAP1, MITF and POT1.39
Prognosis of patients with germline CDKN2A mutations and hereditary melanoma
Patients with germline CDKN2A mutations have an increased risk of developing cutaneous malignant melanoma, with an estimated hazard ratio of 31 in an Australian context. Cust and colleagues’ 2011 study estimated that 20 per cent of Australian CDKN2A mutation carriers will be diagnosed with melanoma by age 50 and 52 per cent by age 80.46 By contrast, the Melanoma Genetics Consortium 2002 study projected a lifetime risk of melanoma in CDKN2A mutation carriers of 91 per cent for those living in Australia.37
There has been some suggestion that CDKN2A-mutant cutaneous malignant melanomas tend to be less invasive and more commonly of the superficial spreading and lentigo maligna subtypes compared to their CDKN2A-wildtype counterparts.3,65 For patients with hereditary melanoma, two retrospective studies have identified no statistically significant difference in Breslow thickness of melanoma, overall survival or the likelihood of developing metastatic disease.33,66 In one study, none of the patients with hereditary melanoma had a positive CDKN2A mutation status, although 11/38 had undergone testing.66 In the other, CDKN2A mutation status was not included as a variable.33 Further research comparing prognosis for patients with hereditary melanoma with and without identified genetic mutations is recommended. Helgadottir and colleagues’ recent studies examining this relationship in the Swedish population have found hereditary melanoma patients with germline CDKN2A mutations to have worse survival rates compared to hereditary melanoma patients with wildtype CDKN2A.67,68
A proposed system of classification
The diagnosis and management of patients with FAMMM syndrome is relevant to the clinician who manages melanoma. There is a paucity of clear guidelines on its diagnostic criteria and its relationship to associated but distinct syndromes, such as hereditary melanoma and B-K mole syndrome. The term ‘B-K mole syndrome’ is now considered to be an archaic term and should not be used today. The terms ‘hereditary melanoma’, ‘genetic melanoma’ and ‘familial melanoma’ are also widely used in the literature. Confusingly, such terms are often used interchangeably with FAMMM syndrome.
The primary aim of this review is to clarify and define the relationship between three distinct entities: hereditary melanoma, FAMMM syndrome and germline CDKN2A mutations. A patient with hereditary melanoma is defined as one who meets Leachman and colleagues’ diagnostic criteria exhibited in Table 1.34,39 As per the criteria, patients in areas with a higher incidence of melanoma need to have more primary melanomas, or have more family members with invasive melanomas in order to meet the diagnosis of hereditary melanoma. FAMMM syndrome is defined as per the GARD and Orphanet criteria discussed above.16,17 The diagnosis of patients with inherited germline CDKN2A mutations is based on the results of genetic testing.
Figure 2 demonstrates a novel system of classification that clarifies the relationship between hereditary melanoma, FAMMM syndrome and patients with germline CDKN2A mutations via a Venn diagram schematic. These are three interrelated but distinct conditions. The key points from our classification system are as follows:
-
Patients may present with hereditary melanoma but not fit the diagnostic criteria for FAMMM syndrome. Such patients may or may not be found to have CDKN2A mutations.69
-
Patients may have hereditary melanoma and fit the clinical phenotype of FAMMM syndrome. Such patients may or may not be found to have CDKN2A mutations.
-
Patients with FAMMM syndrome may have germline CDKN2A mutations, but testing negative for such a mutation does not rule out the diagnosis of FAMMM syndrome.
-
Patients who present with many atypical moles but no family history of malignant melanoma do not fit the criteria for hereditary melanoma and therefore do not warrant genetic testing.
-
Patients who present with early-age onset of melanoma (less than age 40) but no family history of melanoma do not fit the criteria for hereditary melanoma and also do not warrant genetic testing.
A management algorithm
The second aim of this article is to propose a management algorithm that differentiates between patients with FAMMM syndrome, hereditary melanoma and those with germline CDKN2A mutations. The management principles for these three interrelated conditions are similar but have subtle differences which are pertinent for patient education and counselling. The algorithm is presented in Table 2 and is based on the classification system proposed in Figure 2. All patients with FAMMM syndrome are included in a subset of patients with hereditary melanoma. As such, patients are divided into four groups.
The clinical management recommendations for patients with a germline CDKN2A mutation (groups 1 and 3 from Table 2) are the same, regardless of whether they fit the FAMMM syndrome phenotype. These include education on skin protection and the importance of regular three to six-monthly skin checks, including scalp, oral and genital mucosa. Suspicious lesions should be reviewed regularly with the use of total body photography and/or dermoscopy and should have lower thresholds for biopsies. Children should start regular skin checks around the time of puberty because of their risk of early onset melanoma.6,39,70,71 No guidelines currently exist on routine screening for pancreatic cancer in this patient population, although some authors have recommended that carriers undergo annual pancreatic screening via endoscopic ultrasonography or magnetic resonance cholangiopancreatography from age 50 onwards.3,63 Carriers are also recommended to stop smoking because of evidence that smoking in CDKN2A carriers is associated with a higher risk of pancreatic, respiratory and upper gastro-intestinal tract cancers.72
Patients from Group 2, who have hereditary melanoma and the clinical phenotype of FAMMM syndrome but who test negative for CDKN2A mutation, should be informed that CDKN2A mutations account for the minority of cases of hereditary melanoma. If they have a particularly strong family history, then further genetic testing could be offered, such as a panel test for mutations in CDK2, BAP1, MITF and POT1.39 However, prior to offering this testing they should be informed that they are likely to test negative because these four mutations, in total, account for only 1–2 per cent of hereditary melanoma cases.64 The counselling and recommended preventative measures for these patients is similar to that of patients in groups 1 and 3, with the exception that they do not need to be counselled on a possible increased risk of pancreatic cancer. Patients should be informed that melanomas in FAMMM syndrome patients can both develop from melanocytic naevi, but also from normal skin.6,73,74 When doing their regular skin checks, they should also pay extra attention to their trunk, where melanomas disproportionately arise in FAMMM patients.75
Patients from Group 4 should also be offered genetic counselling given their strong family history and may be offered further genetic testing for rarer mutations, as with patients from Group 2. If no significant mutation is identified, patients should still undergo high-level skin surveillance, such as regular total body skin checks every six–12 months and education on sun protection. Family members should also still be linked in with a dermatologist for regular skin checks.6 Testing negative for CDKN2A mutations means that patients from groups 2 and 4 need not be concerned with the increased risk of pancreatic and neural system cancers.47,48,76,77 Aside from this, the negative CDKN2A test does not necessarily bring relief, nor greatly alter clinical management.
Conclusion
The diagnosis and management of patients with FAMMM syndrome is relevant to any clinician managing melanoma. However, clear guidelines on its diagnostic criteria and its relationship to associated but distinct conditions are lacking in the existing literature. The article provides diagnostic and treatment guidelines for three related but distinct conditions: FAMMM syndrome, hereditary melanoma and germline CDKN2A mutations. This review synthesises and presents the current evidence from the literature in a structured and comprehensible manner. The relationships and descriptions of these conditions is addressed via a narrative and historical review. We propose a novel system of classification that describes the complex relationship between these three conditions. A management algorithm for these conditions is described, which designates patients with features from these related conditions into four distinct groups. This article should be of use to plastic surgeons, dermatologists and primary care physicians who manage melanoma routinely but only rarely manage patients with hereditary melanoma.
Conflict of interest
The authors have no conflicts of interest to disclose.
Funding declaration
The authors received no financial support for the research, authorship and/or publication of this article.
Revised: December 4, 2021 AEST